



Geochemical characteristics of deep groundwater are essential information for safety assessments for the geological disposal of radioactive wastes [1], and the sequestration of carbon dioxide [2], one of the known greenhouse gases, because groundwater chemistry could affect migration of the species included in disposal wastes. In order to facilitate the smooth advance of the above disposal projects, it is necessary to investigate the geochemical characteristics economically across a wide area extending over several kilometres.
Existing investigations of groundwater chemistry so far have involved drilling a borehole, purging the drilling mud, pumping up the groundwater, sampling it at the surface and conducting analyses in the laboratory. At potential disposal locations (hereinafter referred to as in-situ), the groundwater is generally under high pressure to dissolve gases, i.e. carbon dioxide, and is in a reduced condition. When pumped up to the surface, it could be degassed with depressurisation to increase its pH and it could be oxidised by contact with the atmosphere to increase its oxidation redox potential (ORP) [3]–[6].
In order to procure quality data on the pH and ORP of the deep groundwater, it is recommended to measure them in-situ [7], [8], and some apparatus has been developed for
in-situ groundwater measurement and sampling. One problem is that since in-situ measurement takes longer and is more expensive, it is difficult to set up a network of measurements consisting of many test intervals in boreholes. A realistic solution is considered as follows: (1) perform not only in-situ measurements, but also the existing ones; (2) develop a method for estimating the in-situ pH and ORP using existing data in comparison with in-situ data; (3) estimate the in-situ values of test intervals where in-situ measurements are not conducted; (4) economically obtain data on the in-situ pH and ORP across a wide area.
This paper suggests a method for estimating the in-situ pH and ORP on the basis of existing data and chemical equilibrium analysis. Moreover, as application examples, the following is estimated: distributions of the in-situ pH and ORP at a site in Japan; a predominant redox reaction governing the in-situ groundwater conditions in use of the groundwater database of Japan.
In order to understand the evolution mechanism of groundwater chemistry, it is convenient to calculate speciation of elements in some environments with a thermodynamic code for geochemical modelling. The geochemical code enables calculations of species activities, concentrations and saturation indices in water on the basis of the mass balance law and the mass action law with a thermodynamic database that includes mass action constants. This paper employs one of the open codes, called PHREEQC [9], developed by the U.S. Geological Survey to estimate in-situ pH and ORP. There are several codes for geochemical modelling other than PHREEQC, which are detailed in the following websites [10].
Since the details of PHREEQC were presented by Parkhurst and Appelo (1999) [9], only a summary of PHREEQC is given here. It is designed to perform a wide variety of low-temperature aqueous geochemical calculations on the basis of an ion-association aqueous model. In order to estimate the in-situ water conditions on the basis of the existing surface data, of the many geochemical calculation capabilities of PHREEQC, speciation and batch reactions with gas at equilibrium are focused on here. Liquid phase interactions with the surrounding solid phase are considered later in the section of RESULT AND DISCUSSION. It uses the mole balance Equation (1), the mass action Equations (2), (3), and the activity coefficient expression including the Davies Equation (4) or the extended Debye-Huckel Equation (5) [11] to calculate the activities, concentrations and saturation indices of the species in solution.
The mole balance equation of an element m is expressed:
where Naq is the number of aqueous species, and Ng is the number of gas-phase species. The moles of each species in the system are represented by ni for aqueous species and ng for gaseous species. The moles of element m per mole of each species are represented by bm, i for aqueous species and bm, g for gaseous species.
The mass action equations can lead to the total moles of an aqueous species i and a gaseous species g:
where n is the moles, K is the mass action constant, am is the activity of master species m, Maq is the total number of aqueous master species, cm is the stoichiometric coefficient of master species m, Waq is the mass of solvent water in an aqueous solution, γ is the activity coefficient, Ngas is the total moles of gas, Ptotal is the total pressure, and subscript i, g represents a solutions species and a gaseous species, respectively.
Activity coefficient γ of aqueous species i is defined with the Davies Equation (4) or the extended Debye-Huckel Equation (5):
where zi is the ionic charge of aqueous species i, μ is the ionic strength of solution, A and B are constants dependent only on temperature, and ai0 and bi are ion-specific parameters fitted from mean-salt activity-coefficient data.
The initial input to PHREEQC was the following analysis data on the groundwater pumped up to the surface: the temperature, pressure (1 atm), pH, ORP, main species concentrations, if there were free gases found, the gas/water ratio, and content of each gas. The groundwater conditions under the in-situ pressure and temperature were computed with PHREEQC on the basis of the initial solution. With increasing pressure, the free gases in the solution were expected to be all solved. The equations below the bubble point were expected to differ from those above it according to the presence or dissolution of the gases. In order to estimate the bubble point, a stepwise computation was applied from the surface pressure and temperature conditions to the in-situ one. If the in-situ mineral information was available, the effects of the mineral on pH or ORP were to be considered and added to the simulation result with PHREEQC.
The estimation method for the in-situ water conditions was applied to a geochemical pumping test for validation. It was performed by the Japan Atomic Energy Agency (JAEA) in the deepest borehole drilled in the course of the Horonobe Underground Research Laboratory Project (Horonobe URL project) [12]. The details of the geochemical pumping test are presented by Hokari and Kunimaru [13], and a summary of the test is given here.
Figure 1 represents the location of the Horonobe URL project, which has been implemented by JAEA in Hokkaido, Japan, with the locations of the investigation boreholes and surrounding geology. The test was conducted at approximately 600 m deep intervals in a vertical borehole named HDB-11. Analysis of the geological column of HDB-11 revealed that it consists of a diatomaceous mudstone Koetoi formation from the surface to a depth of 460 m, and a hard shale Wakkanai formation below 460 m.
Location and Geology of Horonobe URL Project
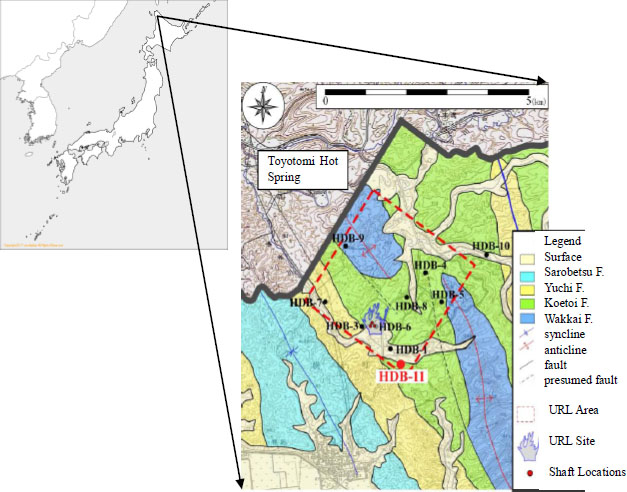
Groundwater at Horonobe site was deduced from the results of some investigations to have evolved as follows: Whereas in the vicinity of the surface, a fresh groundwater of meteoric water origin with a fewer solute contents prevailed, there was a saline groundwater of sea water origin with more solutes in the depths; The present groundwater around the site has been formed by mixing of the above two end waters [12].
Prior to the geochemical pumping test, a hydraulic pumping test was conducted at the same interval of 606 ~ 644 m depth. The following hydraulic properties were obtained:
The geochemical test consisted of pumping the groundwater, monitoring of physical-chemical parameters including water pressure (only in-situ), pH, temperature, dissolved oxygen (DO), electrical conductivity (EC) and ORP, and in-situ water sampling which had the capability of maintaining the in-situ water pressure with stainless steel containers. The parameters were monitored both in-situ and on the surface except for the pressure. Figure 2 presents an outline of the geochemical pumping test apparatus.
Schematic Drawing of the Geochemical Pumping Test Apparatus
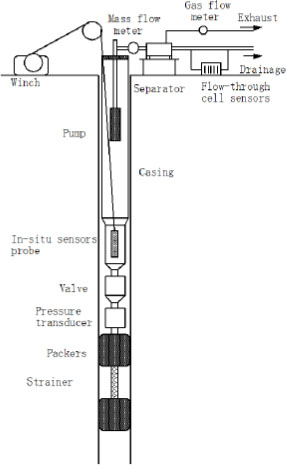
In-situ measurements were made using an OCEAN SEVEN 303 constructed by IDRONAUT, and the surface measurements used WM-50EG for pH, EC and temperature, IM-55G for ORP (Pt electrode) and DO-55G for DO, all made by Toa DKK and installed in the flow-through cell.
The geochemical test procedure is summarised as follows: 1) installation of the pumping apparatus at the test interval in the borehole, 2) packer inflation, 3) pore pressure measurement, 4) installation of in-situ sensor probe, 5) installation of the pump and wellhead assembly, 6) installation of the flow-through cell sensors on the surface, 7) pumping and groundwater monitoring, 8) in-situ groundwater sampling after removal of the in-situ sensors and the pump, 8) six repetitions of the monitoring and water sampling, 9) packer deflation.
Some measurements of the physical-chemical parameters in-situ and on the surface are shown in Figure 3. The horizontal axis represents the ratio of pumped-up groundwater volume to the test interval one of 0.73 m3, and the vertical axis shows pH, EC and ORP_SHE, respectively. ORP_SHE indicates an ORP value relative to the standard hydrogen electrode which was converted from the ORP measurement. EC measurements were corrected and adjusted to 25 °C [14].
Monitoring Result of pH, EC and ORP
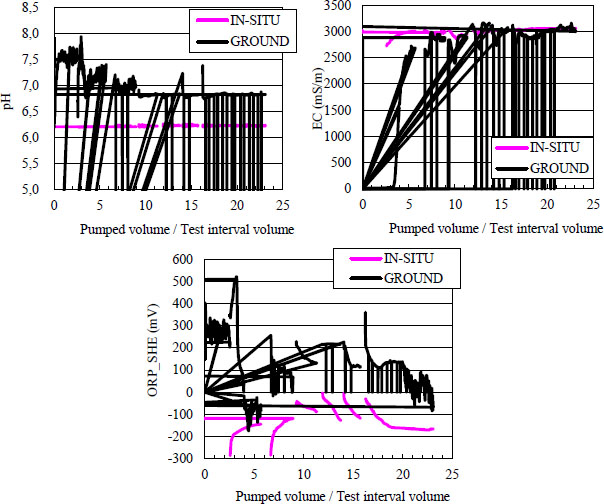
IN-SITU and GROUND in the legends for Figure 3 show measurements monitored in-situ and on the surface, respectively. The pumping procedure included six sets of pumping and monitoring periods and a bottle sampling, the final set of which was the longest. Since the in-situ sensor probe was retrieved from the borehole during the bottle sampling, the measurement curves were intermittent in Figure 3. It is observed that the in-situ measurements differed from the surface ones in pH and ORP, whereas the in-situ EC measurement was in good agreement with that at the surface.
The pore pressure was measured as approximately 6.0 MPa at an initial equilibrium and was stably maintained at 5.8 MPa during pumping. The in-situ temperature was stable and measured approximately 35 °C during the period of the test over around 20 days. DO was not detected in-situ, but a trace amount was detected at the surface.
The flow-through cell sensors were set between a separator and a drain, and measured the groundwater before any contact with the atmosphere. Iwatsuki et al. [6] argued that since the flow cell includes free gases released from the groundwater, which is supposed to be higher than the atmosphere in pressure, it would be less possible to oxidise the groundwater owing to the air intrusion. Grenthe et al. [3] and Gascoyne [5], however, conclude that it is difficult to completely prevent air intrusion into the flow-cell system at the surface. As this pumping test detected DO at the surface, air intrusion could occur in the groundwater pumped up, which could explain the ORP difference between the in-situ and the surface results.
The in-situ groundwater was sampled using high-pressure stainless steel bottles that could maintain the in-situ pressure. Many aqueous species were analysed at the laboratory after the high- pressure bottle samples were depressurised at the site in the air. To analyse a redox couple of Fe2+/Fe3+, the high-pressure water sample was depressurised in an inert atmosphere in the laboratory. In order to measure the gas/water ratios, high-pressure samples were released into a high-pressure vessel of a given volume which had been evacuated so that measurement of the gas pressure may result in a volume of gas being released. The gas contents were analysed using gas chromatography after the gas sampling in the pressure vessel. The aqueous species concentrations and the free gas contents are compiled in Table 1 and Table 2. The gas/water ratios were measured as approximately 1.5. Flame photometry was used for analysis of Na+ and K+. Inductively coupled plasma optical emission spectroscopy was used for Ca2+, Mg2+, Mn2+ and dissolved Si. Absorptiometry was used for NH4+, Fe2+, Fe3+ and S2−. Ion chromatography was used for Cl−, SO4 2− F−, Br−, I− and NO3-. Titration was used for HCO3− and CO32−.
Contents of the groundwater aqueous species
Cation Concentration [mg/L] |
||||||||
|---|---|---|---|---|---|---|---|---|
Na+ |
K+ |
Ca2+ |
Mg2+ |
Mn2+ |
Fe2+ |
Fe3+ |
NH4+ |
Si |
6600 |
140 |
250 |
170 |
0.01 |
2.3 |
<0.05 |
200 |
27 |
Anion Concentration [mg/L] |
||||||||
|---|---|---|---|---|---|---|---|---|
Cl− |
HCO3− |
CO32− |
SO42− |
S2− |
F− |
Br− |
I− |
NO3− |
10000 |
2200 |
0 |
<0.2 |
<0.1 |
0.1 |
78 |
29 |
0.1 |
Free gas contents
O2 |
N2 |
CO2 |
Ar |
CH4 |
C2H6 |
H2S |
|---|---|---|---|---|---|---|
[%] |
[%] |
[%] |
[%] |
[%] |
[%] |
[ppm] |
0.0 |
0.0 |
26.3 |
0.04 |
73.6 |
0.015 |
0.0 |
The stable in-situ pressure during the pumping test implied that no gases would be released from the in-situ groundwater. The in-situ groundwater would release one and a half times the volume of gas as the water on the surface, which consists of 75% CH4 and 25% CO2. This could lead to differences in the pH and ORP between in-situ and surface samples.
According to Figure 3, the pH measurements were stable both in-situ and on the surface. Whereas the in-situ ORP measurement was observed to be stable near the end of the pumping test, the surface measurement was found not to reach a steady state, but to be around −50 ~ −150 mV. The pH and ORP measurements and the PHREEQC computation estimates were as follows:
pH measurements on the surface and in-situ |
6.80 |
6.20 |
pH estimates on the surface and in-situ |
6.80 |
6.29 |
ORP measurements on the surface and in-situ (mV) |
−50 ~ −150 |
−166 |
ORP estimates on the surface and in-situ (mV) |
−198 |
−210 |
As for pH, the surface estimate was in good agreement with the measurement and there was a difference of 0.1 between the in-situ measurement and the estimate. As for ORP, since the surface measurement did not attain a steady state, it was not useful to compare the surface measurement with the estimate. The ORP in-situ estimate indicated a more reduced state than the measurement by 45 mV.
The HDB-11 borehole investigation conducted a rock core analysis as well as the pumping tests, so in-situ mineral information may be available [15]. Mineral effects on groundwater conditions are considered here and added to the PHREEQC computation results.
The groundwater pH would vary according to a change in the carbonate acid equilibrium caused by CO2 degassing with depressurisation. The deep groundwater is expected to be in equilibrium with rock forming minerals in the surrounding formations due to very prolonged residence. The pH would also be governed by carbonate mineral equilibrium such as calcite in-situ, which was identified by the core analysis. A reaction of carbonic acid and water and a pH equation are given in Equation (6). A reaction of calcite dissolution into the water and a pH equation are given in Equation (7). Here, K represents a mass action constant, the activity of an ion is denoted by square brackets, i.e. [Ca2+], and subscript (aq) presents the aqueous species. Two values of log K in each equation correspond to those on the surface and under in-situ conditions.
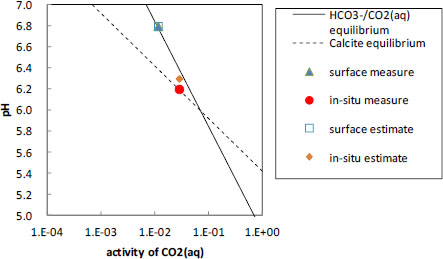
Since PHREEQC computed the above species activities in Equations (6) and (7), the relationship curves between CO2(aq) and pH are given with the measurements and the estimates of pH in Figure 4. A carbonate equilibrium curve is calculated on the surface condition of 15 °C, and a calcite curve on the in-situ condition of 35 °C. The surface pH values are plotted on the carbonate acid equilibrium curve, which is close to the in-situ estimate, and the in-situ measurement is plotted on the calcite equilibrium. The in-situ estimate is actually plotted on the carbonate acid equilibrium curve of 35 °C. It is interpreted that whereas the pH of the groundwater pumped up to the surface will be governed by the carbonate acid equilibrium, the in-situ pH will be governed by the calcite equilibrium. PHREEQC is found to simulate the pH values of the groundwater on the basis of the carbonate acid equilibrium. It is understood that a consideration of the in-situ mineral reaction added to the computation result by PHREEQC would produce a more accurate estimate of the in-situ pH.
Analytical result of the pH measures and estimates
While assuming redox reactions including the aqueous species analysed and the minerals identified [16], [17], the ORP of each reaction in Figure 5 is calculated thermodynamically by the Nernst equation with standard potential, E0, on the basis of the activities of the PHREEQC computation results. Figure 5 shows the calculated ORP-pH relationships with the ORP measurements and the PHREEQC estimates. Although sulphate and sulphide were not detected in the groundwater, pyrite and gypsum were identified from the core analysis. Sulphate and sulphide species are expected to be contained in the water at levels below the detection limit for this analysis of 0.1 mg/L, respectively. The ORP values in Figure 5 are calculated on the assumption that the groundwater should include total sulphur of 1 × 10−7 M, which is approximately a tenth of the detection limit. Some curves running on and near the measurements represent that the in-situ redox condition is governed by the sulphide/sulphate redox reaction or the goethite/siderite reaction, and is also strongly affected by the pyrite/sulphate reaction. This suggests that in-situ, the sulphate/sulphide reaction occurs predominantly after or at the same time as the sulphide supply due to dissolution of the pyrite and/or the iron-containing minerals of pyrite, siderite and ferric oxyhydroxide such as goethite are in equilibrium with each other. ORP behaviours vary widely according to the reactions involved as seen in Figure 5. Here, they could be divided into three patterns, one of which shows higher ORP values than the estimates and the measurements, one of which is lower than those values, and the other of which falls between them. The in-situ ORP is re-calculated on the basis of the latter type of reactions, because they are closest to the in-situ and surface measurements. The result is as follows:
Analytical result of the ORP measurements and estimates
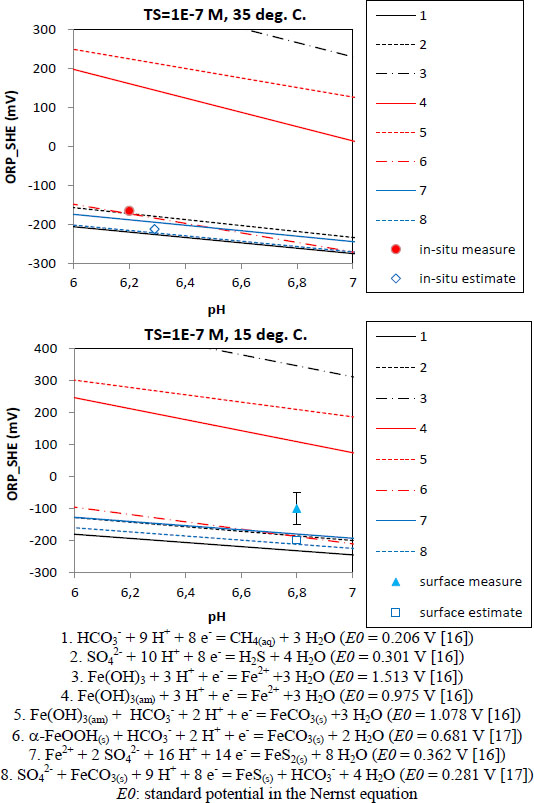
It is understood that a consideration of the in-situ mineral reactions could produce more accurate ORP estimates on the basis of the computation results by PHREEQC.
The estimation results for pH and ORP are compiled with the measurements obtained by the geochemical pumping test in Table 3. The geochemical equilibrium calculations could estimate the in-situ pH and ORP within a margin of error of approximately 0.1 and 50 mV on the basis of the analytical data on the groundwater and the free gases obtained from the pumping test. Furthermore, it is confirmed that as carbonate minerals, such as calcite, affecting the pH were identified in the rock core analysis, a consideration of the in-situ calcite reaction could allow a more accurate estimation of the in-situ pH through use of the activities of aqueous CO2 and Ca2+ computed by PHREEQC. It is also confirmed that consideration of the in-situ redox reactions of the minerals such as pyrite and siderite, which were identified in the rock core analysis, could allow for more accurate estimation of the in-situ ORP in use of the activities of the species concerning the reactions computed by PHREEQC.
Estimation results
Parameter |
Location |
Measurement |
1st Estimate1) |
2nd Estimate2) |
|---|---|---|---|---|
pH |
Surface |
6.80 |
6.80 |
|
In-situ |
6.20 |
6.29 |
6.20 |
|
ORP |
Surface |
−50~−150* | −198 |
|
In-situ |
−166 |
−210 |
−173~−189 |
1) Estimates computed by PHREEQC
2) Estimates calculated on PHREEQC results and mineral reaction
* Provisional value as the measurement reached no steady state
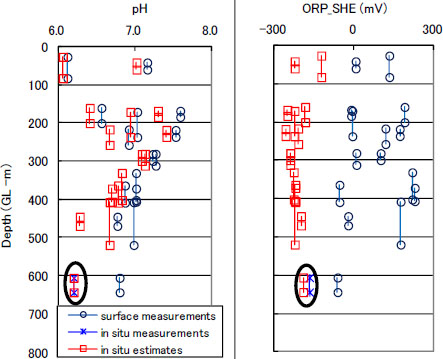
This estimation method is next applied to the several existing pumping tests that were conducted at the Horonobe site [18]. Figure 6 illustrates the depth distributions of the in-situ pH and ORP estimates at the site [19]. The in-situ pH estimates are more acidic than the surface measurements and have a tendency towards acidity to approximately 6.2 with depth. It is coincident with the pumping test results that more CO2 gas was released from the deeper groundwater pumped up to the surface. The in-situ ORP estimates represent a more reduced condition than the surface measurements, and show stable reduction of approximately -200 mV at depths of below some 200 m. This is in agreement with a general tendency that the underground environment is reductive and stable in Japan [17].
An estimation example of the in-situ pH and ORP distributions
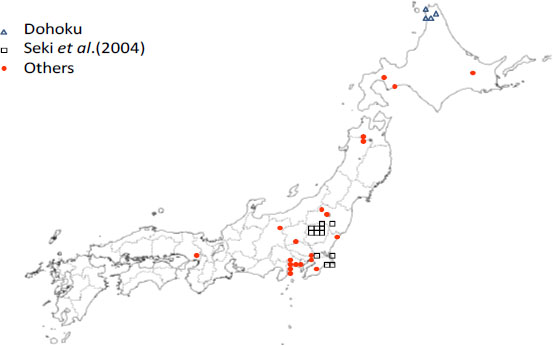
Another application of the method is to groundwater data [21], and the database in Japan [22] for analysis of the in-situ conditions governing redox reactions. Approximately 70 points are selected to satisfy the PHREEQC input data conditions. Figure 7 illustrates the locations of the wells including the Dohoku area (black solid circle), Eastern Kanto district with Seki et al. data [21] (blue solid circle) and other areas (red solid circle). The Dohoku area includes the JAEA boreholes, petroleum wells and hot spring wells. Seki et al. data all comes from hot spring wells. The other area includes natural gas wells, coal mine boreholes, observation wells and hot spring wells. According to the intervals of the wells at which the groundwater was pumped up, the geology is quite different as follows: mudstone, shale, siltstone, sandstone, green tuff, tuffaceous breccia, welded tuff, granite, granodiorite, rhyolite and andesite. Redox reactions involve elements contained in the rocks and the groundwater, which could vary in valence states depending on the redox condition. At first, the elements of Fe, Mn, S and C are selected as redox relevant ones in the rocks, because they exist in greater content in the various rocks [23]. The relative contents of the elements are summarised in Table 4.
Locations of the wells for the in-situ redox reaction estimation
Main elements concerning the redox state and relative contents (modified from [23])
Igneous rocks |
Fe > Mn > S > C |
|
|---|---|---|
Sandstone |
Fe > C > S > Mn |
|
Sedimentary rocks |
Shale |
Fe > C > S > Mn |
Carbonates |
C > Fe > S > Mn |
The selected groundwater data seldom includes Mn, and the following redox reactions are assumed with the elements of Fe, S and C in the in-situ groundwater and the rocks.
The predominant reaction of all the above could be revealed with thermodynamic analysis using the Gibbs reaction energy. The energy was calculated for each reaction for each groundwater data under the in-situ temperature and pressure conditions. The probability of each reaction is as follows:
Reaction |
(8) | (9) | (10) | (11) | (12) | (13) | (14) |
| Probability [%] | 0 | 0 | 34 | 1 | 12 | 36 | 17 |
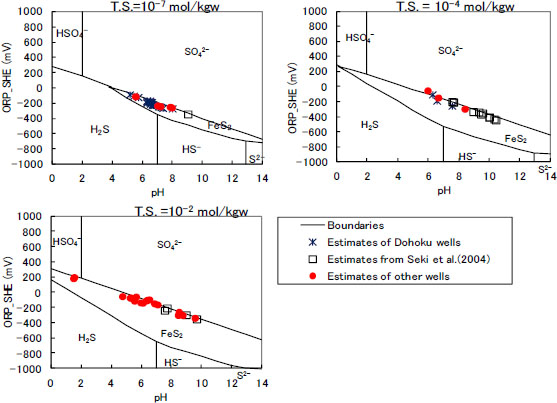
The above probability means, for example, that Reaction (13) is the most likely to occur in 36% of all the data. The sulphate/ferrous sulphide mineral reactions are estimated to be predominant in more than half of the data, the ferrous ion/ferric oxihydroxide reaction predominates in 34% of data, and the siderite/ferric oxihydroxide reaction prevails in 12% . In other words, the redox reactions of the ferrous sulphide minerals are estimated to govern the in-situ groundwater conditions. The in-situ pH and ORP estimates are analysed on the basis of Reaction (13), as shown in Figure 8. Most of the estimates are plotted on an equilibrium curve between pyrite and sulphate. It is deduced from the result that the redox state of the in-situ groundwater could be governed by the pyrite-sulphate reaction in some areas of Japan.
Estimation example of the in-situ redox reaction
This study developed a means of estimating the in-situ pH and ORP, which are very important parameters affecting migration properties in the safety assessment of underground disposal facilities. This was applied to a geochemical pumping test for validation. The following application examples were also shown: When applied to several pumping tests in a given area, it could estimate distributions of the in-situ groundwater pH and ORP in the area; applied to a range of data on deep groundwater in a database of Japan, it could help estimate the in-situ redox reactions governing the groundwater conditions.
Since in-situ pH and ORP measurement is very expensive and time-consuming in the case of borehole investigations, this method could be utilised as follows: (1) The cost and time for the groundwater investigation is expected to be reduced by means of in-situ pH and ORP estimation by using data obtained from existing pumping tests; (2) A safety assessment prior to the site investigation is expected to be performed by means of the in-situ pH and ORP estimation on the basis of the existing groundwater database.
| A | - | the constant dependent only on temperature for the Davies Equation and the extended Debye-Huckel Equation |
| ai0 | [m] | the ion-specific parameter fitted from mean-salt activity-coefficient data for the extended Debye-Huckel Equation |
| am | - | the activity of master species m |
| [a] | - | the activity of species a |
| B | [/m] | the constant dependent only on temperature for the extended Debye-Huckel Equation |
| bi | - | the ion-specific parameter fitted from mean-salt activity-coefficient data for the extended Debye-Huckel Equation |
| bm | - | the moles of element m per mole of species |
| cm | - | the stoichiometric coefficient of master species m |
| E0 | [V] | the standard potential in the Nernst equation |
| K | - | the mass action constant |
| Maq | - | the total number of aqueous master species m |
| m | - | element |
| - | master species | |
| Naq | - | the number of aqueous species in the system |
| Ng | - | the number of gas-phase species in the system |
| Ngas | [mol] | the total moles of gas |
| N | [mol] | the moles of species in the system |
| Ptotal | [atm] | the total pressure |
| Waq | [kg] | the mass of solvent water in an aqueous solution |
| z | - | the ionic charge |
| Greek Letters | ||
| γ | - | the activity coefficient |
| μ | - | the ionic strength of solution |
| Subscripts | ||
| i | a solution species | |
| g | a gaseous species | |
dissolved oxygen
electrical conductivity
Japan Atomic Energy Agency
oxidation-redox potential
an ORP value relative to the standard hydrogen electrode which was converted from the ORP measurement
the Underground Rock Laboratory
- ,
, NUMO , 2004 - ,
Lessons from the First Japanese Pilot Projects on Saline Aquifer CO2 Storage ,J. Geography , Vol. 117 (No. 4), :734-7522008, https://doi.org/10.5026/jgeography.117.734, pp - ,
Redox potentials and redox reactions in deep groundwater systems ,Chem. Geol. , Vol. 98 (No. 1-2), :131-1501992, https://doi.org/10.1016/0009-2541(92)90095-M, pp - ,
Evolution of Redox Conditions and Groundwater Composition in Recharge-Discharge Environments on the Canadian Shield ,Hydrogeol. J. , Vol. Vol. 5 (No. 3), :4-181997, https://doi.org/10.1007/s100400050253, pp - ,
Hydrogeochemistry, groundwater ages and sources of salts in a granitic batholiths on the Canadian Shield, south-eastern Manitoba ,Applied Geochem. , Vol. 19 , :519-5602004, https://doi.org/10.1016/S0883-2927(03)00155-0 - ,
A notice for measuring physicochemical parameters (pH, ORP) of deep groundwater ,J. Jap. Assoc. Groundwater Hydrol. , Vol. Vol. 51 (No. 3), :205-2142009, https://doi.org/10.5917/jagh.51.205, pp - ,
Development of an apparatus to measure groundwater qualities in situ and to sample groundwater using boreholes ,Environ. Geol. , Vol. 32 (No. 1), :17-221997, https://doi.org/10.1007/s002540050189, pp - ,
An Appropriate Manner of Hydrochemical Investigation of Groundwater Using Deep Borehole ,J. Jap. Soc. Eng. Geol. , Vol. Vol. 46 (No. 4), :232-2362005, https://doi.org/10.5110/jjseg.46.232, pp - , User’s Guide to PHREEQC (Version 2), Water-Resources Invest Rep., 1999
- ,
, http://www.ggsd.com/, Geotechnical & geoenvironmental software directory, [Accessed: 27-June-2008] - ,
WATEQ, A computer program for calculating chemical equilibria of natural waters ,J. Research, U.S. Geological Survey , Vol. 2 , :233-2741974 - ,
Horonobe Underground Research Laboratory Project Investigation Report for the 2005 Fiscal Year ,Japan Atomic Energy Agency , 2006 - ,
Estimation of In-situ Deep Groundwater Conditions Based upon Chemical Equilibrium Analysis ,Tech. Research Report Inst.Technol. Shimizu Corp. , Vol. 87 , :77-862010 - , , 1999
- ,
Mineral and chemical composition of rock core and surface gas composition in Horonobe Underground Research Laboratory Project (Phase 1) ,JAEA-Data/Code 2007-022, JAEA , 2008 - , Aqueous environmental geochemistry, Prentice Hall Inc., 1997
- ,
Aquatic chemistry: chemical equilibria and rates in natural waters -3rd ed. ,A Wiley-interscience publication, John Wiley & Sons, Inc. , 1996 - ,
Analysis of Ground Water from Boreholes ,River Water and Precipitation in the Underground Research Laboratory Project, JAEA-Data/Code 2007-015, JAEA , 2007 - ,
Estimation of distributions of the in-situ groundwater physic-chemical parameters ,Proceedings of the 2008 Autumn Meeting of Japanese Association of Groundwater Hydrology, JAGH , 294-299, Nov. 20-22, 2008pp - ,
The draft second progress report on research and development of HLW disposal in Japan - H12 project to establish technical basis for HLW disposal in Japan - Supporting report 1 - Geological environment in Japan ,JNC-TN 1400 99-011, JNC , 1999 - ,
Discharged water from deep wells in the eastern Kanto region, - the relationship between water quality and underground geology ,J. Balneological Society of Japan , Vol. 54 (No.1), :1-242004, pp - ,
Information of geological features of the Japanese Islands ,JNC TN7450 2002-003, Japan Atomic Energy Agency , 2003 - , Study and interpretation of the chemical characteristics of natural water, 1970